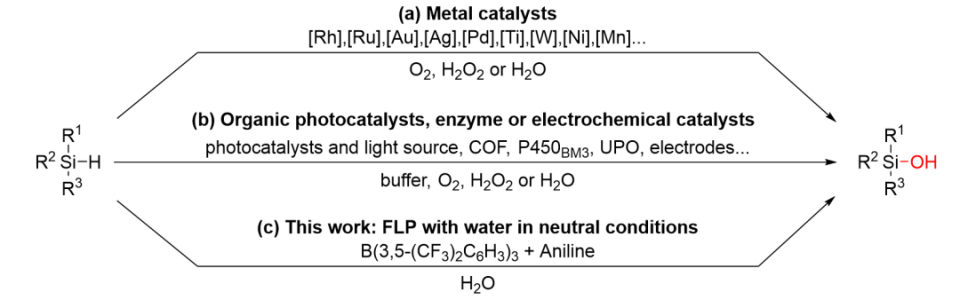
近日,吉林大学超分子结构与材料国家重点实验室张越涛教授课题组与南京大学理论与计算化学研究所黎书华教授课题组进行合作,开发了硅烷氧化可控合成硅醇的催化体系,相关成果以标题为“Frustrated Lewis pairs-promoted organocatalytic transformation of hydrosilanes into silanols with water oxidant”发表在Journal of the American Chemical Society上。硅醇是一类具有重要价值的有机化合物。在材料化学领域中,它常常作为含硅聚合物的构筑原料;在有机合成领域,硅醇又是一些含硅生物活性分子或药物分子的常用硅源,也是一些有机反应中的导向基团、催化剂或配体。目前合成硅醇的方法大多是过渡金属催化,以氧气、过氧化氢或水作为氧化剂(图1a)。然而,非金属催化体系在该反应中应用极少,只有一些光催化、电催化或酶催化的例子,这些反应体系依赖光源、电源或者必须在缓冲溶液中进行,降低了体系的操作性和实用性(图1b)。B(C6F5)3作为有机合成中常用的路易斯酸,其可以催化硅烷氧化生成硅醇,生成的硅醇又会在B(C6F5)3的催化作用下立即与体系中的硅烷发生脱氢偶联反应生成硅醚。因此,通过简单方便的反应装置、绿色的水氧化剂以及硼非金属催化剂,实现硅醇专一性生成的方法是值得探究的。基于张越涛教授课题组在受阻路易斯酸碱对(FLP)化学的研究背景,利用B(3,5-(CF3)2C6H3)3/苯胺FLP作为催化剂,水作为氧化剂,我们实现了可控的硅烷氧化反应,以高产率和高选择性合成了一系列带有不同取代基的硅醇,并避免了副产物硅醚的生成(图1c)。基于此,将该氧化反应和B(C6F5)3催化的脱氢偶联反应相结合,精准合成了几种序列可控的硅醚低聚物,这是一个将小分子反应应用于聚合物控制性合成的很好的实例。
 我们选择二苯基甲基硅烷(1a)为底物,对FLP催化氧化体系的反应条件进行筛选(表1)。首先,选择常用的B(C6F5)3为路易斯酸催化剂,N,N-二乙基苯胺作为路易斯碱,水作为氧化剂,在100°C下2小时后底物仅转化了53%,得到51%的硅醇产物2a以及2%的硅醚副产物3a(表1,条目1)。随后我们尝试了酸性稍强于B(C6F5)3但位阻较小的B(3,5-(CF3)2C6H3)3来催化反应,得到了很好的反应效果:反应2小时后目标产物2a以96%的产率生成(表1,条目5)。体系置于25°C下则不能进行转化(表1,条目8)。此外,若没有路易斯碱的参与,硅烷则会以93%的产率生成硅醚(表1,条目9)。最终在一系列的筛选下,确定了反应的最优条件:2 mol%的B(3,5-(CF3)2C6H3)3和4 mol%的PhNEt2作为催化剂,20 μL水作为氧化剂,0.2 mmol的硅烷在100°C下反应2小时。制备级实验以92%的高分离产率实现了硅醇的合成,证明了该体系的实际应用前景。
我们选择二苯基甲基硅烷(1a)为底物,对FLP催化氧化体系的反应条件进行筛选(表1)。首先,选择常用的B(C6F5)3为路易斯酸催化剂,N,N-二乙基苯胺作为路易斯碱,水作为氧化剂,在100°C下2小时后底物仅转化了53%,得到51%的硅醇产物2a以及2%的硅醚副产物3a(表1,条目1)。随后我们尝试了酸性稍强于B(C6F5)3但位阻较小的B(3,5-(CF3)2C6H3)3来催化反应,得到了很好的反应效果:反应2小时后目标产物2a以96%的产率生成(表1,条目5)。体系置于25°C下则不能进行转化(表1,条目8)。此外,若没有路易斯碱的参与,硅烷则会以93%的产率生成硅醚(表1,条目9)。最终在一系列的筛选下,确定了反应的最优条件:2 mol%的B(3,5-(CF3)2C6H3)3和4 mol%的PhNEt2作为催化剂,20 μL水作为氧化剂,0.2 mmol的硅烷在100°C下反应2小时。制备级实验以92%的高分离产率实现了硅醇的合成,证明了该体系的实际应用前景。表1 反应条件优化
aReaction conditions: 1a (0.2 mmol), H2O (20 μL), C6D6 (0.6 mL), in air. Conversions and yields were determined by 1H NMR spectroscopy; bin N2; cwithout H2O.
在最优反应条件下,我们探索了适用的硅烷底物的范围(图2)。为了将不同位阻和电子特性的硅烷均以高产率和高选择性氧化为相应的硅醇,本文选择了三种不同的苯胺作为路易斯碱来组成FLP用于催化氧化反应:中等体积的PhNEt2(N1),较小位阻的PhNMe2(N2)和较大位阻的PhNiPr2(N3)。从氧化反应的结果中可以总结出,氧化硅原子附近位阻较大的硅烷底物时,较小位阻的苯胺可以提高反应的效率,但有时会降低反应的选择性。而对于较小位阻的硅烷,例如二甲基苯基硅烷,小位阻的苯胺虽然能达到100%的转化,但反应的控制性差,会生成较多的硅醚副产物,这时选择大位阻苯胺并降低反应温度,即可在保证转化率的同时还能获得较好的选择性。底物中含有不饱和的碳碳双键或三键,以及带有环张力的环丙基以及含有萘基、吲哚基和噻吩基的底物都不会发生其它位的硅氢化反应。此外,当底物中含有两个Si-H键时,两个反应位点都可以得到很好地氧化。但若选择小位阻苯胺催化该反应时会生成硅醚聚合物。受此启发,我们又合成了三官能度和四官能度的硅烷,为我们后续得到一些拓扑结构的含硅低聚物打下了基础。由于该FLP催化氧化体系实现了以前工作中难以实现的可控性,我们设计了一系列的对照实验来探究这一全新的反应机理。首先,根据已报道的工作,我们推测B(3,5-(CF3)2C6H3)3会首先与硅烷底物作用发生Si-H键的活化。向等摩尔比的二苯基甲基氢硅烷(Ph2MeSiH)和三苯基氘代硅烷(Ph3SiD)混合液中加入硼路易斯酸或FLP,在核磁监测下发现二者之间有氢氘交换现象。这证实了硼路易斯酸或FLP对Si-H存在活化作用(图3a)。接下来,向FLP中加入当量的硅烷,在核磁监测中没有发现有峰位移的变化(图3b);而向FLP中加入水,很快观察到了新物种的生成(图3c)。将该物质进行分离纯化,并利用单晶X射线衍射进行了结构表征(图3d)。由结构可知,FLP将水的H-O键进行了异裂,生成了离子对[PhEt2NH][HO-B(3,5-(CF3)2C6H3)3](A)。上述结果说明,FLP对H-O键的活化是优先于Si-H的。为了证明FLP的协同活化作用在该反应体系中的重要性,我们使用B(3,5-(CF3)2C6H3)3和异喹啉组合的路易斯酸碱加合物作为催化剂来催化反应,发现其并不能对水起到活化作用,也不能使氧化反应进行(图3e)。为了更加深入地理解反应机理,我们与南京大学黎书华教授合作,利用DFT计算研究了体系中化合物及中间态的自由能分布(图4)。首先,B(3,5-(CF3)2C6H3)3路易斯酸、PhNEt2路易斯碱与体系中的H2O迅速生成稳定的离子对Int1。该离子对可以对硅烷底物以协同的方式进行活化,经历SN2的TS1/2过渡态后,生成路易斯酸与羟基氧配位的加合物Int2以及路易斯碱,并释放氢气(图4,虚线)。该过程经历了19.2 kcal/mol的能垒,在100°C加热的条件下是可以进行的,这也与室温下该反应不发生的现象是相符的。在过量水存在的情况下,中间体可能会以氢键的形式结合更多的水,生成更稳定的结构(Int1’,图4,实线)并跨过稍高的能垒(24.7 kcal/mol)。然而该能垒在加热的反应条件下也是容易实现的。而在不加入路易斯碱的情况下,B(3,5-(CF3)2C6H3)3路易斯酸单独催化硅烷氧化则会在室温下快速生成硅醚(表1,条目9)。对于这一过程进行DFT计算也可以得到合理的解释:类似于FLP参与的过程,路易斯酸单独催化硅烷同样经历了SN2过渡态生成硅醇,能垒为11.0 kcal/mol(图5)。一旦体系内生成了硅醇,它作为亲核试剂进攻硅烷,会经历相似的SN2过程生成硅醚,能垒为8.4 kcal/mol(图6)。由于生成硅醚的能垒低于生成硅醇的能垒,在路易斯酸单独催化的条件下,氧化反应无法停留在硅醇产物上,而是更容易进一步生成硅醚,这也是与实验结果相符的。对比FLP催化的体系,由于FLP对水的优先、快速的活化,避免了路易斯酸单独对硅烷的活化,从而避免了硅醚副产物的生成,这体现出了受阻路易斯酸碱对协同活化的优势。基于上述实验,我们提出了可能的反应机理(图7,ArF=3,5-(CF3)2C6H3)。首先B(3,5-(CF3)2C6H3)3/苯胺FLP协同活化H2O生成离子对[PhEt2NH][HOB(3,5-(CF3)2C6H3)3](A)。在加热的条件下离子对A对硅烷的Si-H键进行活化,以SN2机理进行氧化并脱去氢气。生成的加合物会与体系中的水反应,得到目标产物硅醇,并完成离子对A的再生。聚合物的精准合成方法近年来受到越来越多的关注,我们将本章开发的FLP氧化体系与B(C6F5)3催化的脱氢偶联反应相结合,通过控制加料顺序,实现了序列可控硅醚低聚物的精准合成。如图8a所示,每一步反应都有80%以上的产率,五步反应即可以60%总产率得到含有三个硅原子的硅醇,随后合成含有六个硅原子的硅醚低聚物。利用双官能度的硅烷还可实现序列向两端的延伸(图8b)。利用平面结构的三官能度以及四面体中心结构的四官能度硅烷实现了具有空间拓扑结构硅醚低聚物的合成(图8c和d)。体系利用水作为氧化剂,生成氢气作为唯一的副产物,是将小分子反应向聚合物合成的成功扩展案例。总之,本章开发了一个非金属FLP催化、水氧化的体系,高选择性、高产率地实现了硅烷氧化生成硅醇的反应。反应成功的关键在于FLP对水的优先活化,不仅促使了氧化反应的发生,也避免了硅醚副产物的生成。基于此本文还开发了精准合成硅醚低聚物的方法,以较高的产率和严格的控制性合成了几种空间拓扑结构硅醚低聚物,实现了小分子反应向聚合物合成的成功扩展。该成果发表在Journal of the American Chemical Society上。吉林大学化学学院博士研究生谢馥伃、博士研究生张苏韬和南京大学化学化工学院博士研究生杨沫为论文共同第一作者。吉林大学化学学院张越涛教授、何江华教授和南京大学化学化工学院黎书华教授为论文通讯作者。此研究得到国家自然科学基金等资助支持。